¿Es contagiosa la enfermedad de Alzheimer?
![]()

En la actualidad se conocen más de cincuenta demencias resultantes de enfermedades neurodegenerativas, y la enfermedad de Alzheimer es la más extendida. Se estima que cerca de 35 millones de personas la sufren en el mundo, y que el número de enfermos podría alcanzar los 115 millones de personas en los próximos 30 años.
Aunque en algunos casos —pocos— el alzhéimer puede deberse a un problema genético, el factor de riesgo principal es el envejecimiento. Así pues, es lógico que la prevalencia de estas enfermedades guarde relación con el aumento de la esperanza de vida.
No obstante, recientemente se han llevado a cabo trabajos que también apuntan a que, en determinadas condiciones, la enfermedad de Alzheimer podría transmitirse entre personas. Es cierto que su mecanismo recuerda el de las enfermedades producidas por priones, como la enfermedad de Creutzfeldt-Jakob, de la que se produjeron decenas de casos como consecuencia de la crisis de las vacas locas. A continuación ofrecemos algunas explicaciones.
La enfermedad de Alzheimer, un problema de proteínas
Desde un punto de vista sintomático, el alzhéimer se caracteriza por un deterioro drástico de las facultades psíquicas y físicas debido a la muerte de las neuronas cerebrales. Cuando se analizan los cerebros de los pacientes fallecidos, se observa la presencia de dos tipos de depósitos de proteínas.
Las proteínas son moléculas de gran tamaño constituidas por una secuencia de moléculas más pequeñas denominadas aminoácidos. Se trata de componentes esenciales de la vida: las células y los tejidos de los seres vivos contienen miles de proteínas, que cumplen funciones variadas y específicas (hormonas, enzimas, proteínas de estructura como el colágeno, la tubulina, que constituye el “esqueleto” de las células y les da su forma, etc.). En la enfermedad de Alzheimer, algunas de estas proteínas se vuelven anormales y se acumulan.
El primer tipo de depósitos proteicos encontrados en el cerebro de pacientes con esta demencia contiene una proteína denominada “Tau” (del inglés Tubulin-associated unit). En condiciones normales, una de sus funciones es estabilizar la estructura de las neuronas. En la enfermedad de Alzheimer, Tau se modifica y deja de cumplir su función. Las neuronas se degeneran, mientras que las proteínas anormales se agregan unas a otras y se acumulan en las células nerviosas.
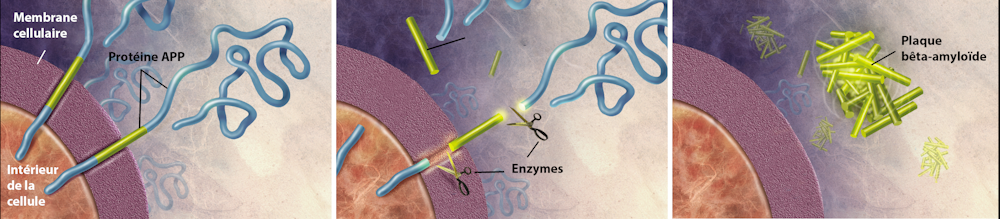
El péptido Aβ se produce a partir de un corte en la secuencia de aminoácidos de una gran proteína denominada APP o precursor proteico amiloide. Esta proteína se sitúa en la superficie de las neuronas e interviene sobre todo en su crecimiento, su supervivencia y su reparación.
En condiciones normales, los péptidos Aβ se eliminan, pero en la enfermedad de Alzheimer se acumulan en el exterior de las células nerviosas, en forma de placas amiloides, también denominadas placas seniles. Estos depósitos se encuentran, asimismo, alrededor de los capilares sanguíneos del cerebro y pueden ser causa de microhemorragias cerebrales denominadas angiopatías amiloides cerebrales.
Wikimedia, CC BY
Proteínas capaces de contaminar otras
El aspecto más destacable de los mecanismos que dan lugar al alzhéimer es que la neurodegenerescencia no se deriva de una simple acumulación pasiva de proteínas.
En realidad, las proteínas implicadas cambian de forma, lo que modifica su acción a escala celular. Cabe señalar que la función de una proteína depende generalmente de su forma (que, a su vez, depende en gran medida de la secuencia de los aminoácidos que la compone). Este cambio de morfología es lo que confiere al péptido Aβ propiedades totalmente diferentes de las de su forma normal. Al ser capaz de autoagregarse, puede formar los depósitos de fibras amiloides que, probablemente, causan la muerte de las neuronas.
Pero aún hay más: los investigadores han demostrado que las formas tóxicas son capaces de obligar a sus álter ego normales a imitarlas y a adoptar a su vez una forma patógena. Este fenómeno, denominado “autorreplicativo”, explica cómo una célula enferma que produce la forma tóxica del péptido puede “contaminar” la célula vecina.
Este contagio de una célula a otra también explica por qué, durante la evolución de la enfermedad de Alzheimer, la propagación de las lesiones cerebrales se extiende de forma progresiva a todo el cerebro siguiendo un esquema bien determinado que se observa en todos los pacientes.
Mecanismos similares a los de los priones
Este proceso autorreplicativo es similar al que se observa en el caso de otra enfermedad neurodegenerativa, la enfermedad de Creutzfeldt-Jakob. Esta se debe a la propagación en el seno del cerebro de un agente patógeno muy particular: el prion.
Aunque no es una bacteria, ni un parásito, ni un virus, ni un hongo, el prion es, sin embargo, transmisible. El descubrimiento de estas “partículas proteicas infecciosas” (el acrónimo prion procede del inglés proteinacious infectious particule) ha sido objeto de un amplio debate y ha obligado a los investigadores a crear un nuevo concepto, el de “agentes transmisibles no convencionales”. Al contrario de lo que ocurre con los otros agentes patógenos, los priones están desprovistos de genoma (no tienen ADN ni ARN) y se componen exclusivamente de una única proteína.
Como sucede con las proteínas implicadas en la enfermedad de Alzheimer, las células producen de forma natural una versión “normal” del prion. Esta versión cumpliría numerosas funciones biológicas, pero todavía no se conocen bien sus diversas utilidades. También posee la propiedad de replegarse y agregarse para formar partículas infecciosas. En su forma infecciosa, los priones son capaces de infectar a un nuevo individuo tras la ingesta de determinados tejidos contaminados, o a través de la sangre, por ejemplo.
La gran resistencia de los priones a los procesos de destrucción clásicos ha dado lugar a varias crisis económico-sanitarias de gran alcance, como la crisis de las vacas locas en los años ochenta y noventa o el escándalo de la hormona del crecimiento contaminada.
¿Es contagiosa?
Los procesos de agregación del péptido Aβ y de la proteína Tau presentan semejanzas con el proceso observado en los priones. Así pues, ¿es posible que la enfermedad pueda transmitirse entre personas, según un mecanismo idéntico al del prion? Diversos grupos de científicos han intentado responder a esta pregunta.
De forma experimental, varios equipos de investigadores han podido inducir la proliferación de agregados de péptidos Aβ en animales de laboratorio. Por otra parte, más recientemente, se han llevado a cabo diferentes trabajos que apuntan a la existencia de casos de transmisión iatrogénica del péptido Aβ patógeno, lo que ha dado lugar a angiopatías amiloides cerebrales. Las hormonas de crecimiento producidas, en particular, antes de 1977 no solo habrían estado contaminadas por el prion, sino también por el péptido Aβ, y habrían podido intervenir en el desarrollo de la enfermedad de Alzheimer.
Paralelamente a la publicación de estos trabajos, se ha detallado de manera especial otra población llamada “de riesgo”, la de los pacientes que han recibido un trasplante de duramadre. Esta fina membrana fibrosa que protege el cerebro podía extraerse de cadáveres y utilizarse como “gasa” tras la realización de operaciones neuroquirúrgicas invasivas. Esta práctica se prohibió en Francia en 1994, pues los trasplantes de duramadre dieron lugar a casos de transmisión iatrogénica del prion humano.
Se llevaron a cabo tres estudios (suizo, japonés e internacional) que mostraron respectivamente que el 71,4%, el 81% y el 61,5% de los pacientes que recibieron este tipo de trasplante desarrollaron posteriormente angiopatías amiloides cerebrales. Aunque no es posible aportar la prueba formal de la contaminación por causa de los trasplantes, la localización de las lesiones y de los depósitos proteicos indica con claridad que el trasplante es la causa del cambio de forma y de la agregación de los péptidos Aβ del receptor.
Un estudio también ha señalado que los instrumentos quirúrgicos utilizados en neurocirugía tal vez podrían constituir a veces una fuente de contaminación, aunque el riesgo es probablemente muy bajo. No obstante, los autores de estos trabajos proponen que se mejoren los procedimientos de esterilización.
¿Es una enfermedad producida por priones?
Desde un punto de vista mecanístico, queda claro que la enfermedad de Alzheimer se parece a las enfermedades producidas por priones. Si nos atenemos a la definición estricta de la palabra prion —partícula proteica infecciosa—, la enfermedad de Alzheimer debería estar incluida, puesto que ha quedado demostrado el carácter transmisible de los ensamblajes proteicos tóxicos que la causan, al menos de forma experimental.
No obstante, de acuerdo con los resultados de investigación, el concepto de prion se ha ampliado: se ha revelado la existencia de diferentes cepas de priones así como su capacidad para “mutar” y adaptarse a su nuevo huésped. En este aspecto, las enfermedades producidas por priones se diferencian de la enfermedad de Alzheimer. Así pues, de acuerdo con el nivel de conocimientos actual, parece más justo calificar esta última como “enfermedad de tipo prion” o “de amiloides infecciosos”. O ampliar el término de agente transmisible no convencional a los ensamblajes proteicos causantes de la enfermedad de Alzheimer.![]()
Sobre la autora: Angélique Igel-Egalon es ingeniera de investigación del grupo de macroagregados proteicos y enfermedades priónicas del Inra
Este artículo fue publicado originalmente en The Conversation. Artículo original.