Un modelo teórico para acelerar el descubrimiento de nuevos fármacos
Este texto es una colaboración del Cuaderno de Cultura Científica con Next
Uno de los principales problemas y costes a los que se enfrenta la investigación de nuevos fármacos es comprobar que una nueva molécula candidata como mínimo interactúa adecuadamente con su diana terapéutica. Si el que una molécula tenga un efecto beneficioso es abrir una cerradura con una llave, lo complicado es fabricar (sintetizar químicamente, lo que no siempre es sencillo) cientos de miles de llaves diferentes y después comprobar que, como mínimo, encajan en la cerradura (lo que no es baladí experimentalmente). Por eso disponer de un modelo teórico que permitiese predecir cómo se unirá una molécula a una diana es algo muy necesario para la industria farmacéutica y para la humanidad en su conjunto. Pensemos en lo que acortaría el trabajo para encontrar nuevos fármacos para nuevas epidemias o nuevos antibióticos para los que las bacterias aun no hubiesen desarrollado resistencia. Ahora el esfuerzo conjunto de un numeroso grupo de investigadores de las Universidades de Columbia, Yale y California en Irvine y de empresas privadas parece que habría encontrado ese modelo buscado. Publican sus resultados en el Journal of the American Chemical Society.
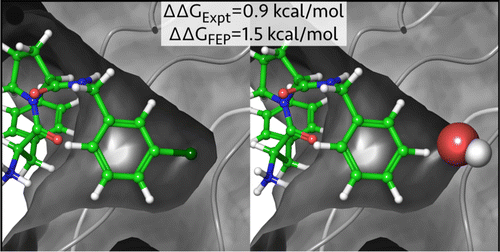
Muchos fármacos (ligandos) funcionan enlazándose al centro de unión de una proteína (receptor) bloqueándolo o modificando su función. Predecir lo bien que una molécula candidata se enlazará implica calcular el cambio de energía libre en el proceso. En esto pueden influir muchos factores: las interacciones electrostáticas y de van der Waals, los enlaces por puente de hidrógeno, los efectos entrópicos, cambios en la interacción con el disolvente (solvatación) y varios más. Este tipo de cálculos se han hecho anteriormente con éxito pero de forma muy específica; lo que es difícil es encontrar métodos que funcionen para distintos tipos de interacciones ligando-receptor y que no requieran cantidades ingentes de capacidad de computación.
Lo que los investigadores han hecho ahora es encontrar una forma mejor de calcular los campos de fuerza que modelan las interacciones interatómicas. Para ello se han basado en un trabajo previo desarrollado en la Universidad de Yale a finales de los años ochenta por William Jorgensen y sus colaboradores llamado Potenciales Optimizados para Simulaciones Líquidas (OPLS por sus siglas en inglés). De ahí que al nuevo método lo hayan bautizado como OPLS 2.1.
OPLS 2.1 también simplifica los algoritmos que se emplean para comprobar las diferentes conformaciones posibles de la pareja receptor-ligando, reduciendo así la cantidad de datos iniciales que el usuario tiene que introducir. La facilidad de uso es fundamental si realmente quiere ser una herramienta útil en el descubrimiento de nuevos fármacos.
Los investigadores han comprobado su método con 200 ligandos diferentes frente a 8 receptores, lo que cubre un amplio espectro de sistemas biológicos: desde enzimas del ciclo celular a la trombina que coagula la sangre. Alrededor del 78% de los compuestos que OPLS 2.1 predecía que tendrían un buen encaje se encontró que era así experimentalmente; pero se alcanzó el 92% de acierto con las moléculas en las que el enlace sería débil.
Con todo hay que tener en cuenta que los cálculos de variación de energía libre nos dirán si una reacción se ve favorecida termodinámicamente, pero el equilibrio de las reacciones es tan sensible a estos cambios que las pequeñas discrepancias con la realidad que puede tener un cálculo pueden ser la diferencia que hay entre lo que funciona teóricamente y lo que es inútil en la práctica. Por tanto, como es lógico, los cálculos en un ordenador no pueden sustituir a la experimentación, pero sí ayudar a dirigir recursos escasos, financieros y temporales, hacia las moléculas más prometedoras.
Referencia:
Lingle Wang et al (2015) Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field J. Am. Chem. Soc.,137 (7), pp 2695–2703 DOI: 10.1021/ja512751q
Sobre el autor: César Tomé López es divulgador científico y editor de Mapping Ignorance