«El bacteriófago ø29 como sistema modelo en biología molecular» por Margarita Salas

Este texto de Margarita Salas apareció originalmente en el número 2 de la revista CIC Network (2007) y lo reproducimos en su integridad por su interés.
Después de tres años en el laboratorio de Severo Ochoa, Eladio Viñuela y yo decidimos regresar a España en 1967 para tratar de desarrollar la Biología Molecular que habíamos aprendido con Ochoa. Una decisión importante era elegir el proyecto de trabajo. El verano anterior habíamos seguido un curso sobre virus bacterianos en los laboratorios de Cold Spring Harbor. Precisamente, el trabajo con los virus bacterianos había dado lugar al nacimiento de la Genética Molecular en la década de los 50. Eladio y yo elegimos un fago como sistema modelo para estudiarlo a nivel molecular. Pusimos dos condiciones al fago a elegir: una, que fuese pequeño pero complejo desde el punto de vista estructural y, otra, que fuese poco conocido pues sabíamos que el comienzo en España llevaría tiempo y no queríamos empezar con un tema muy competitivo. La otra decisión que tomamos fue empezar a trabajar juntos, pues siempre sería más fácil nuestro comienzo si uníamos y complementábamos nuestros esfuerzos.
Después de repasar la literatura que existía sobre distintos fagos, Eladio encontró un trabajo publicado por el laboratorio de Dwight Anderson en el que describían la morfología de la partícula y el tamaño del DNA del fago ø29 de Bacillus subtilis. Este fago tenía las características que estábamos buscando: tamaño pequeño, morfología compleja (ver figura 1) y muy poco conocido. Puesto que en aquella época no había financiación estatal para hacer investigación en España, presentamos un proyecto a la Jane Coffin Childs Memorial Fund for Medical Research utilizando el fago ø29 como modelo para estudiar su morfogénesis y los mecanismos de transferencia de la información genética. Gracias a esta ayuda, pudimos empezar nuestro trabajo en Madrid en el Centro de Investigaciones Biológicas del Consejo Superior de Investigaciones Científicas. El Director del Instituto Marañón del Centro, José Luis Rodríguez Candela, nos ofreció un laboratorio espacioso para empezar nuestra investigación. Recuerdo la emoción que nos produjo nuestro primer experimento que consistió en crecer un cultivo de la bacteria Bacillus amyloliquefaciens, infectarlo con el fago ø29 y ver que en 40-50 minutos a 37°C la bacteria lisaba. Esto suponía que ya teníamos el sistema para iniciar nuestro trabajo.
Pocos meses después de nuestra vuelta a España se convocaron las primeras becas de “Formación de Personal Investigador” por lo que pudimos seleccionar a nuestros primeros estudiantes de doctorado. El primero fue Enrique Méndez quien caracterizó las proteínas estructurales de la partícula viral utilizando la técnica de electroforesis en geles de poliacrilamida en presencia de dodecil sulfato sódico que había desarrollado Eladio en Nueva York. Después de Enrique, llegaron Jesús Ávila y Antonio Talavera. Jesús purificó la RNA polimerasa de B. subtilis, implicada en la transcripción del DNA de ø29, y demostró que estaba formada por varias subunidades equivalentes a β’, β, σ y α del enzima de Escherichia coli. Tengo que confesar lo orgullosos que nos sentimos cuando nos aceptaron en la revista Nature el primer trabajo que describía las características de la RNA polimerasa de B. subtilis. Por su parte, Antonio Talavera aisló los primeros mutantes letales condicionales de ø29, en su caso mutantes sensibles a temperatura (ts). Posteriormente, Felipe Moreno aisló mutantes sensibles a supresor (sus) de ø29.
En paralelo a la obtención de mutantes ts y sus de ø29 en nuestro laboratorio, el grupo de Anderson había aislado otra colección de mutantes ts y sus. Decidimos juntar las dos colecciones de mutantes y construimos un mapa genético lineal con un total de 17 genes que numeramos del 1 al 17 de acuerdo con su colocación en el mapa de izquierda a derecha. Por otra parte, Marta R. Inciarte y José M. Lázaro construyeron un mapa físico relativo al mapa genético utilizando la nucleasa de restricción EcoRI. Esta fue la primera vez que se utilizó en España un enzima de restricción.
Mediante el uso de los mutantes sus disponibles, Ana Camacho, Fernando Jiménez, José L. Carrascosa y Javier de la Torre caracterizaron la ruta morfogenética para el ensamblaje de las proteínas y la encapsidación del DNA necesarios para la construcción de una partícula viral.
Se suponía que el DNA de ø29 era lineal pero Juan Ortín lo aislaba en forma circular o formando concatémeros que se convertían en DNA lineal de longitud unidad por tratamiento con enzimas proteolíticos. Esto significaba que la circularización y concatemerización del DNA estaba mediada por proteína. Este trabajo se publicó en Nature New Biology. Posteriormente, caracterizamos una proteína codificada por ø29 unida covalentemente a los extremos 5’ del DNA del fago que después demostramos que estaba implicada en la iniciación de la replicación del DNA de ø29. A esta proteína, producto del gen 3 viral, la llamamos proteína terminal (TP). La Figura 2 muestra una fotografía al microscopio electrónico, tomada por José M. Sogo, que muestra una molécula de DNA lineal de ø29 con la TP en los extremos.
José M. Hermoso, José M. Sogo, Marta R. Inciarte y Javier Corral demostraron la existencia de un control temporal en la transcripción del DNA de ø29, mapearon los promotores y demostraron que después de la infección con un mutante sus 4 la transcripción tardía no tenía lugar. Uno de los objetivos fue tratar de caracterizar la proteína producto del gen 4, lo cual era difícil ya que la proteína no parecía sintetizarse en cantidades altas y la infección con ø29 no inhibe la síntesis de proteínas de la bacteria. Afortunadamente, en la década de los 70 pudimos disponer de las herramientas de la ingeniería genética por lo que fue posible clonar genes y sobreproducir proteínas.
Otro cambio importante tuvo lugar a finales de los 70. En 1977 nos trasladamos al Centro de Biología Molecular “Severo Ochoa”. El trabajo de Eladio en la parte científica y el de Javier Corral y Juan A. Manzanares en la técnica hicieron posible la existencia y calidad del Centro.
A continuación describiré el trabajo que realizamos en el Centro de Biología Molecular “Severo Ochoa”, centrado principalmente en el estudio de los mecanismos de control de la transcripción del DNA de ø29 y en la replicación de su DNA que utiliza la proteína terminal como iniciadora (“primer”).
Control de la transcripción del DNA de ø29
Al comienzo de la infección con ø29 sólo se expresan los genes implicados en la replicación del DNA y en la regulación de la transcripción (genes tempranos). Los genes que codifican a las proteínas estructurales del fago y a las proteínas implicadas en morfogénesis y en la lisis celular se expresan posteriormente (genes tardíos). Localizamos los sitios de iniciación de la transcripción e identificamos la secuencia de los cuatro promotores tempranos principales (A1, A2c, A2b y C2) y la del único promotor tardío (A3). Los promotores A2c y A2b son responsables de la expresión de los genes implicados en la replicación del DNA (6, 5, 3, 2, 1 y 56) y en el control de la transcripción (4 y 6). El promotor C2, localizado a la derecha del genoma, es el primero que se transcribe y dirige la expresión de los genes 17 y 16.7, ambos implicados en la inyección del DNA y en su replicación. Previo a la inyección, el fago se adsorbe específicamente a la pared bacteriana a través de los apéndices del cuello, como demostró Nieves Villanueva. El promotor tardío A3, localizado próximo al promotor temprano A2b, es responsable de la expresión de los genes 7 a 16 que codifican a proteínas estructurales, morfogenéticas y de lisis.
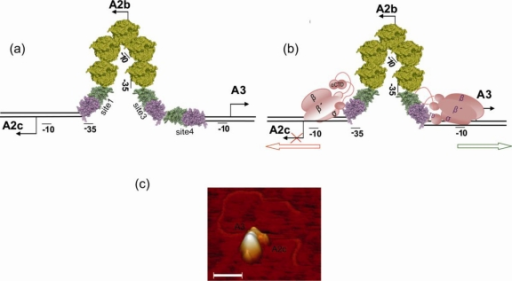
La caracterización del producto del gen 4 fue posible debido, por una parte, a la secuenciación del gen y, por otra, a su clonación en E. coli bajo el control del promotor PL del fago λ y sobreproducción de la proteína. La purificación de la p4 y la puesta a punto de un sistema de transcripción in vitro permitió determinar que la proteína p4 es un activador del promotor tardío A3 y, a su vez, reprime al promotor temprano A2b, ya que excluye a la RNA polimerasa del mismo. El promotor A2c, localizado a la izquierda del A2b, también es reprimido por p4 mediante un mecanismo que implica la unión simultánea de p4 y RNA polimerasa, evitando el escape de la polimerasa del promotor. Posteriormente, demostramos que la expresión de los promotores A2b, A2c y A3 es regulada por la proteína p6, además de la p4.
La estructura cristalina de p4 sola y en complejo con un DNA que incluye el sitio de unión del promotor A3, se determinó en colaboración con el grupo de Miquel Coll. La proteína p4 contiene un nuevo motivo de unión al DNA que consiste en una estructura en forma de garfio en cada extremo N-terminal de un homodímero. Los dos garfios entran en el surco mayor de la doble hélice estableciendo contactos específicos con una guanina. La curvatura del DNA permite que los dos garfios contacten los dos surcos mayores que se encuentran separados tres vueltas de hélice. Los resultados de mutagénesis dirigida indicaron que los únicos contactos específicos son entre la arginina 6 de la p4 y la G de la repetición invertida del sitio de unión, siendo el resto de los contactos con el esqueleto fosfato del DNA.
Recientemente, Ana Camacho demostró que, además, existe reconocimiento específico de secuencia de DNA a través de lectura indirecta de series de adeninas (“A-tracts”).
Replicación del DNA de ø29 iniciada por la proteína terminal
La secuencia de los extremos del DNA de ø29 mostró la existencia de una repetición terminal invertida de seis nucleótidos (AAAGTA). La TP está unida a los extremos del DNA por un enlace fosfoester entre el grupo OH del residuo serina 232 y 5’AMP.
El análisis por microscopía electrónica de los intermedios replicativos sintetizados en B. subtilis infectado con ø29, indicó que la replicación empieza en cualquiera de ilustresmolde, daba lugar a la síntesis de TP-DNA de longitud unidad de un modo muy procesivo. Cuando se usó como molde DNA de M13 utilizando un oligonucleótido como “primer” la DNA polimerasa de ø29 sintetizó procesivamente un DNA de más de 70 kb, indicando que es una polimerasa muy procesiva capaz de producir desplazamiento de banda sin necesidad de proteínas accesorias. Debido a estas propiedades y a la actividad correctora de pruebas, la DNA polimerasa de ø29 está siendo utilizada comercialmente como herramienta para la amplificación isotérmica de DNA circular y de DNA genómico lineal.
Mecanismo de deslizamiento hacia atrás (“sliding-back”) para iniciar la replicación del DNA de ø29
La replicación de fragmentos de DNA de ø29 llevó a Juan Méndez a descubrir que la iniciación no ocurre en el extremo 3’terminal (T) sino en la segunda base (T) desde el extremo 3’. Una vez formado el complejo de iniciación, dirigido por el segundo núcleotido, el complejo TP-dAMP se desliza hacia atrás para recuperar la T 3’ terminal. La iniciación interna también ocurre en otros fagos relacionados con ø29 como Nf y GA-1, así como los fagos Cp1 y PRD1 y los adenovirus.
Estudios de estructura-función de la DNA polimerasa y proteína terminal de ø29
Cuando empezamos esta parte del trabajo no disponíamos de la estructura tridimensional de la DNA polimerasa de ø29. La comparaciónlos dos extremos del DNA, de un modo no simultáneo, y transcurre hacia el otro extremo por desplazamiento de banda.
Cuando Miguel A. Peñalva incubó extractos de B. subtilis infectado con ø29 con [α32P] dATP en presencia de TP-DNA de ø29 como molde, encontró una proteína marcada con la movilidad electroforética de la TP. Este producto era sensible a álcali indicando la formación de un complejo covalente TP-dAMP. Posteriormente, demostramos que los genes 2 y 3 virales son necesarios para la formación de dicho complejo. Se clonaron los genes 2 y 3 y las proteínas se sobreprodujeron y purificaron. Luis Blanco demostró que la proteína p2, además de catalizar la reacción de iniciación (formación de complejo TP-dAMP), tiene actividad DNA polimerasa. Además, tiene actividad exonucleasa 3’-5’ sobre DNA de banda simple, con las propiedades que se esperan de un enzima implicada en corrección de pruebas.
La TP y DNA polimerasa purificadas, en presencia de TP-DNA como de la secuencia de aminoácidos de DNA polimerasas procarióticas y eucarióticas llevó a Luis Blanco y Antonio Bernad al hallazgo de una serie de motivos conservados. Propusimos que el sitio activo de la exonucleasa 3’-5’ en DNA polimerasas está formado por tres motivos de aminoácidos en el dominio N-terminal que contienen los cuatro grupos carboxilato que unen dos iones metálicos. El análisis de mutantes en cada uno de estos residuos indicó que las mutaciones producían una actividad exonucleasa cien veces menor. Miguel de Vega analizó mutantes en otros residuos que estaban implicados en la unión al DNA de banda simple y a la TP. Por otra parte, mutagénesis dirigida en motivos de aminoácidos conservados en el dominio C-terminal mostró que este dominio está implicado en la polimerización y en la iniciación con TP como “primer”, y se identificaron aminoácidos implicados en la unión a metal y catálisis, unión a DNA, a la TP y a los dNTPs. Fueron muchas las personas implicadas en este trabajo, en particular José Ma. Lázaro quien purificó todos los mutantes de la DNA polimerasa.
Recientemente, se ha determinado la estructura cristalina de la DNA polimerasa de ø29 en colaboración con el grupo de Tom Steitz en la Universidad de Yale. Esta estructura explica su extraordinaria procesividad y capacidad de desplazamiento de banda. Las DNA polimerasas que utilizan TP como “primer” tienen dos inserciones específicas, TPR1 y TPR2. La región TPR1 interacciona con la TP mientras que TPR2 forma una especie de abrazadera que podría conferir la alta procesividad de la polimerasa. Efectivamente, la deleción de la región TPR2 dio lugar a una drástica reducción en la procesividad de la polimerasa y en su capacidad de realizar desplazamiento de banda. También se ha determinado la estructura del heterodímero DNA polimerasa: proteína terminal. La estructura de la TP muestra un dominio (N-terminal) que no interacciona con la DNA polimerasa, otro (intermedio) que interacciona con la polimerasa y un tercer dominio (“priming”) que contiene el residuo de serina 232 que ocupa el mismo sitio de unión en la polimerasa que ocupa el DNA dúplex durante la elongación.
Proteínas virales p6 y p5, esenciales para la replicación del DNA de ø29
La proteína p6 de unión a DNA de banda doble (DBP) es una proteína tipo histona, esencial para la replicación del DNA de ø29 in vivo, muy abundante en B. subtilis infectado por ø29 (~700.000 moléculas/célula). La p6 se une como dímero al DNA de ø29, preferentemente a sus extremos, cada 24 nucleótidos de forma cooperativa formando un complejo nucleoprotéico. Manuel Serrano demostró que la unión de p6 a DNA circular produce superenrrollamiento positivo proponiendo un modelo en el cual una superhélice positiva de DNA se une alrededor de una estructura de p6. Por microscopía electrónica, Crisanto Gutiérrez demostró que el DNA está compactado unas 4 veces en el complejo con p6. La formación del complejo facilitaría la apertura de los extremos del DNA para iniciar la replicación. De hecho, la p6 estimula la formación in vitro del complejo de iniciación TP-dAMP.
La proteína p5 de unión a DNA de banda simple (SSB) es también muy abundante en células infectadas y es esencial para la elongación de la replicación in vivo.
Amplificación in vitro del DNA de ø29
Usando las cuatro proteínas purificadas mencionadas, TP, DNA polimerasa, p6 (DBP) y p5 (SSB) Luis Blanco pudo amplificar in vitro más de 1.000 veces cantidades pequeñas de TP-DNA (0.5 ng) en una hora a 30°C. La infectividad del DNA amplificado fue idéntica a la del DNA de ø29 obtenido de partículas de virus, lo que indica la fidelidad de la amplificación.
Proteínas de membrana implicadas en la replicación el DNA de ø29: proteínas p1 y p16.7
La replicación del DNA de ø29 se reduce considerablemente en B. subtilis infectado con un mutante sus en el gen 1. La proteína p1 se asocia con la membrana bacteriana a través de su extremo C-terminal. Además, la p1 se autoasocia dando lugar a largas láminas bidimensionales e interacciona con la TP in vitro. Estos resultados sugieren que la p1 forma parte de una estructura viral asociada a la membrana que suministra un sitio de anclaje para la maquinaria de replicación del DNA de ø29.
La replicación del DNA de ø29 está retrasada en B. subtilis infectado con un mutante sus 16.7. Wilfried Meijer demostró que p16.7 es una proteína integral de membrana, siendo requeridos los 22 aminoácidos N-terminales para ello. La proteína p16.7 a la que se le ha quitado el dominio transmembrana forma dímeros y multímeros en solución y se une a DNA de banda simple y doble sin especificidad de secuencia, así como a la TP. Daniel Muñoz-Espín demostró que los 70 aminoácidos C-terminales de p16.7 (p16.7C) constituyen la parte funcional de la proteína. En colaboración con los laboratorios de Juan Luis Asensio y Armando Albert se ha determinado la estructura cristalina y en solución de p16.7C, así como la estructura cristalina de un complejo trimérico p16.7C- DNA de doble banda.
Otras proteínas virales implicadas en la replicación del DNA de ø29: proteínas p17 y p56
Cuando se infecta B. subtilis con un mutante sus en el gen 17 la síntesis del DNA viral se reduce. La p17 interacciona con la proteína p6 y estimula la unión de ésta a los extremos del DNA de ø29.
El gen 56 codifica una proteína de 56 aminoácidos cuyo blanco celular es la uracil-DNA glicosilasa (UDG). Gemma Serrano-Heras y Alicia Bravo demostraron que la adición de p56 a extractos de B. subtilis inhibe la actividad UDG celular. La inhibición de la UDG celular por p56 es un mecanismo de defensa desarrollado por ø29 para evitar la acción del proceso de reparación por excisión de bases si se producen residuos de uracilo en los intermedios replicativos de banda simple en el proceso de replicación del DNA de ø29.
Interacciones fago-bacteria en el desarrollo de ø29
Se conocía que el fago ø29 no se desarrollaba eficientemente en B. subtilis capaz de esporular. Wilfried Meijer y Virginia Castilla-Llorente demostraron que el ciclo lítico de ø29 se suprime cuando la bacteria infectada ha iniciado el proceso de esporulación y el genoma del fago infectante se localiza en la espora y permanece “durmiente” hasta la germinación de éste. En este proceso están implicadas dos proteínas celulares. Por una parte, la proteína SpoOJ, implicada en la segregación del cromosoma bacteriano, que a través de los sitios de unión que existen en el DNA de ø29, parece estar implicada en el atrapamiento del genoma de ø29 en la espora. Por otra parte, la proteína SpoOA, que regula la iniciación de la esporulación, se une a varios sitios de unión que existen en el genoma de ø29 y suprime el desarrollo de ø29 reprimiendo los promotores tempranos evitando la activación del promotor tardío. Además, SpoOA es también un inhibidor de la replicación, tanto del DNA de ø29 como del DNA de B. subtilis. Mediante su unión a los orígenes de replicación, SpoOA previene la iniciación de la replicación de ambos genomas.
Comentarios finales
El trabajo realizado en mi laboratorio durante 40 años ha sido un trabajo de investigación básica que ha tratado de desvelar las bases moleculares del desarrollo de ø29. Pero esta investigación básica ha dado lugar a una aplicación biotecnológica importante: el uso de la DNA polimerasa de ø29 para la amplificación del DNA, tanto de DNA circular como de DNA genómico, lineal. Este es un buen ejemplo de cómo haciendo investigación básica se pueden obtener aplicaciones que no se preveían. Este año se cumple el 40 aniversario del comienzo en España de nuestro trabajo con ø29. Quiero resaltar el hecho de que el trabajo realizado en mi laboratorio es el resultado de la dedicación e ideas de las personas que han trabajado en el grupo durante estos 40 años apasionantes. Mi mayor gratitud a todos tanto a los que han sido citados como a los que, por falta de espacio, no he podido citar. Mi agradecimiento también a mis dos maestros, Alberto Sols y Severo Ochoa, quienes me enseñaron no sólo la Bioquímica y la Biología Molecular, respectivamente, sino también su rigor experimental, su entusiasmo y su dedicación a la investigación. Pero especialmente quiero expresar mi agradecimiento a Eladio Viñuela. Juntos iniciamos nuestra aventura de desarrollar la Biología Molecular en España y, si esta aventura ha tenido éxito, se lo debemos a Eladio.

Margarita Salas Falgueras. Presidenta de la Fundación Severo Ochoa. Profesora ad honorem en el Centro de Biología Molecular Severo Ochoa (CSIC-UAM). Ha recibido, entre otros premios, el nombramiento Investigadora Europea (1999) por la UNESCO, el Ramón y Cajal de Investigación y el Rey Jaime I. Miembro de la Real Academia de Ciencias Exactas, Físicas y Naturales, de la Real Academia Española y de la Academia Nacional de Ciencias de EE.UU.
Edición realizada por César Tomé López a partir de materiales suministrados por CIC Network
"El bacteriófago ø29 como si…
[…] Este texto de Margarita Salas apareció originalmente en el número 2 de la revista CIC Network (2007) y lo reproducimos en su integridad por su interés. Después de tres años en el laboratorio de Sev… […]
Reocín se suma a la celebración del Día Mundial de la Mujer y la Niña en la Ciencia – Top Cantabria Fm
[…] El bacteriófago ø29 como sistema modelo en biología molecular» por Margarita Salas. Edición realizada por César Tomé López a partir de materiales suministrados por CIC Network, número 2 (2007) […]