Sobre la predisposición genética a padecer enfermedades (II)

En un artículo anterior hablábamos de cómo se descubren las variantes genéticas (los polimorfismos de un solo nucleótido ‒SNPs‒, ¿recuerdas?) que podemos relacionar estadísticamente con diversas enfermedades o trastornos mediante estudios de asociación del genoma completo (GWAS, Genome-wide association study). Es importante dejar claro que estas asociaciones nos señalan algunos cambios en el ADN que aumentan la probabilidad de que padezcamos una patología, sin que ello signifique que la vayamos a sufrir necesariamente.
Entonces ya apuntábamos a un problema de estas correlaciones SNP-patología: salvo excepciones, no indican de forma clara la causa concreta de la enfermedad. Sabemos que un nucleótido concreto en una posición específica del genoma aumenta la predisposición a padecer un trastorno determinado, pero poco conocemos sobre los mecanismos celulares y rutas metabólicas que llevan de una variación concreta del ADN a la enfermedad. Y esto es importante si queremos investigar tratamientos efectivos.
En algunos casos sí que podemos deducir las causas: si hay un cambio en la secuencia de un gen, este cambio puede afectar a la estructura de la proteína resultante y, según las funciones de esta, podemos tratar de inferir los mecanismos que llevan a sufrir el trastorno. Pero esto sólo ocurre en un 5-10% de los SNPs estudiados y asociados a patologías: el resto de variantes genéticas relacionadas con enfermedades a través de GWAS se encuentran en zonas del genoma que no codifican proteínas (en el mal denominado ADN basura).
¿Qué ocurre en el resto de casos? ¿Cómo puede afectar una variante genética, un pequeño cambio en la secuencia de ADN, al funcionamiento de la célula si no se encuentra directamente implicada en la construcción de proteínas? Pues a través de otros mecanismos, principalmente mediante la regulación de la expresión génica.
Expresión génica
Aunque la secuencia de ADN de todas nuestras células es la misma (salvo mutaciones puntuales), resulta evidente que existen muchos tipos distintos de células: las células que forman los músculos son distintas a las del hígado o el páncreas, a las epiteliales o a las neuronas. Esto se debe a que no se «activan» los mismos genes ni con la misma intensidad en las distintas células.
Recordemos que los genes son secuencias de ADN que contienen la información para fabricar una proteína. Para ello, previamente se transcriben a ARN mediante la acción de la enzima ARN polimerasa, se eliminan fragmentos no codificantes de su interior (los denominados intrones) y las secuencias resultantes (denominadas ARNm) se traducen a proteínas. Un cambio en la secuencia de ADN que define un gen puede conllevar que la proteína se fabrique mal y que aparezca una enfermedad grave.
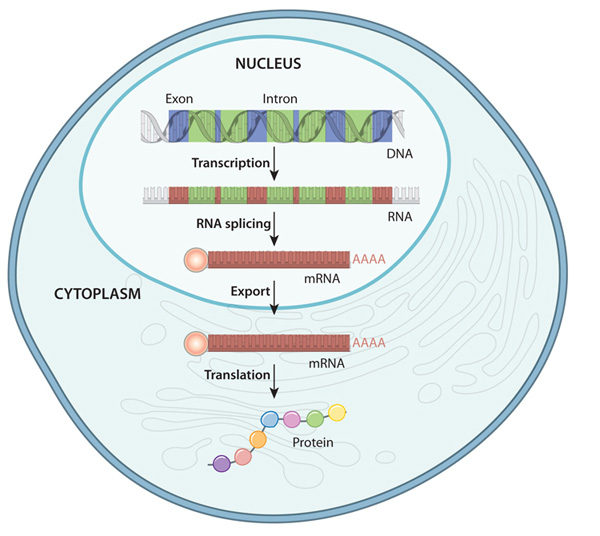
Así pues, un gen contiene la información necesaria para fabricar una proteína, pero no sobre cuándo empezar este proceso, cuánta cantidad de proteína es necesaria y en qué tipo de células se debe llevar a cabo. De esta regulación de la expresión génica, se encargan factores externos, diversos actores en el interior de la célula y, en particular, otras secuencias cromosómicas. Un ejemplo sería la propia secuencia que marca el inicio de transcripción donde se une la enzima ARN polimerasa (promotor). Otros fragmentos de ADN van a actuar como potenciadores de la transcripción o represores. A grandes rasgos, estos fragmentos (que no tienen por qué encontrarse cerca del gen que controlan) regulan el plegamiento de la cadena de ADN como lugares de unión de otros complejos enzimáticos, permitiendo un mejor o peor acceso de la ARN polimerasa al inicio de la transcripción. Una variación en estas secuencias no influye en cómo se construye la proteína, sino en qué condiciones se va a fabricar y en qué magnitud.
Podemos medir la expresión de un gen en un tejido concreto estimando qué cantidad de ARNm se ha fabricado, lo que se conoce como transcriptoma. Avances recientes en secuenciación genética han permitido obtener transcriptomas con una eficacia elevada.
Expression quantitative trait loci
Así pues, resumiendo, podemos utilizar técnicas avanzadas de secuenciación masiva para obtener tanto el genoma de un conjunto de individuos como su transcriptoma. De hecho, empezamos a disponer de grandes bases de datos con ambos datos asociados a patologías y rasgos concretos de los donantes de muestras. Analizando estas bases de datos con potentes herramientas computacionales y estadísticas podemos relacionar variaciones concretas del genoma (en particular, SNPs) en regiones no codificantes, con cambios en la expresión génica. A estas regiones del ADN cuyos cambios afectan a la expresión génica de uno o más genes se les denomina eQTLs (expression quantitative trait loci). Vamos a utilizar esta definición con frecuencia a lo largo de este artículo, así que vamos a explicar con más detalle qué significa.
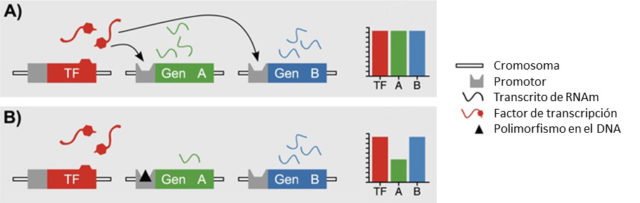
Un eQTL es una posición concreta del genoma que, en función del nucleótido que se encuentre en ella, va a influir en la predisponibilidad a poseer algún rasgo o padecer enfermedad concreta. Pero no lo va a hacer modificando la estructura de la proteína codificada por un gen, sino alterando la expresión génica. Es decir, las personas que presenten en su genoma un eQTL determinado van a fabricar proteínas de forma correcta, pero en una cantidad distinta de la adecuada para el funcionamiento adecuado de la célula, pudiendo dar lugar a algún trastorno determinado o rasgo específico.
En la figura anterior, un factor de transcripción se une a los promotores de los genes A y B, dando como resultado una cierta cantidad de proteína (A). Si hay una variación en el promotor del gen A (indicada con un triángulo negro en la figura) la cantidad de proteína fabricada disminuye.
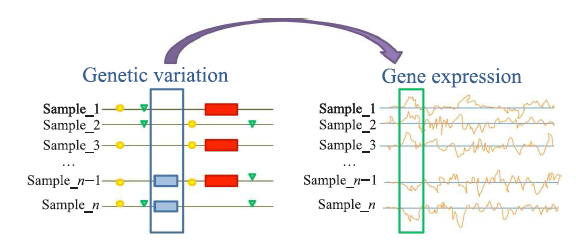
¿Cómo se localizan eQTLs en el genoma? Primero, se obtienen los SNPs de cada uno de los participantes en el experimento como se explicó en el anterior artículo. En la siguiente figura se toman como ejemplo dos polimorfismos de los cromosomas 4 y 14, marcados como SNP1 y SNP2. Recuerda que un SNP varía de individuo a individuo, aunque en este ejemplo sólo consideraremos dos variantes posibles.
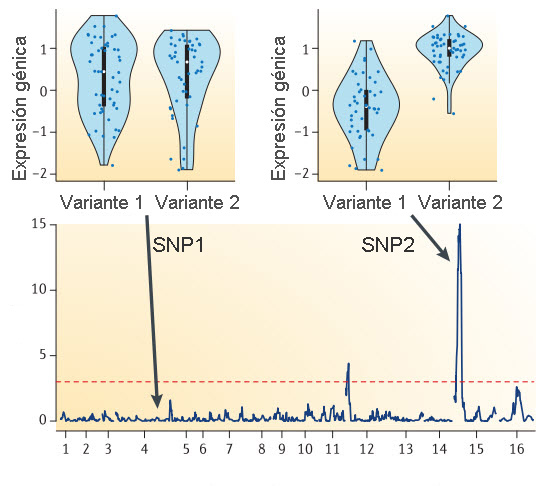
A continuación se obtiene una muestra del tejido de interés (recuerda que la expresión génica depende del tipo concreto de células) y se mide la cantidad de ARNm fabricado de distintos genes (con técnicas como RNA-seq). Podemos representar para cada variante de un SNP la cantidad de ARNm expresada de un gen concreto (en la figura, los puntos negros de las gráficas). Se observa que en el primer SNP no hay apenas variación en el nivel de expresión según se posea una variante u otra (fíjate en las formas de las distribuciones de puntos, destacadas en fondo azul), mientras que en el segundo hay una mayor expresión en la variante 2. Por lo tanto, diríamos que el SNP2 tiene asociados cambios destacables de expresión génica según la variante implicada, por lo que sería un eQTL.
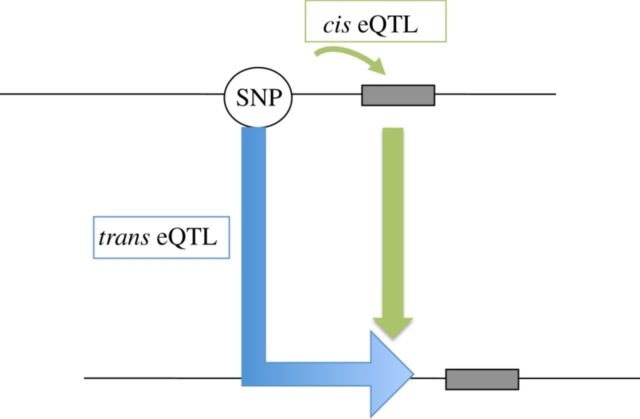
Con este tipo de estudios se han podido localizar un gran número de eQTLs en nuestro genoma. Muchas de estas zonas influyen en la expresión génica por encontrarse cerca del lugar donde se inicia la transcripción del gen: se conocen como cis-eQTL. Hay que tener en cuenta que «cerca» puede significar hasta un millón de nucleótidos de distancia entre el SNP y el inicio del gen. Otras variaciones, sin embargo, son capaces de modificar la expresión de un gen situado a mucha distancia del SNP implicado, e incluso en un cromosoma distinto: son los denominados trans-eQTL.
Empezábamos este artículo comentando que conocer que una variante genética esté asociada a una enfermedad no implica que podamos entender cuál es el mecanismo subyacente, lo cual no nos da pistas de cómo tratar el problema. Si descubrimos que esta variante afecta a la expresión de un gen determinado, y conocemos su función celular, sí que podemos investigar nuevos tratamientos.
Pongamos un ejemplo concreto. Recientemente se descubrió un SNP asociado a un menor riesgo de padecer un infarto de miocardio. Se comprobó que la variante menos común de este SNP (la T destacada en azul en la siguiente figura) promueve la expresión de un gen en el hígado que produce sortilina. Estudios posteriores con ratones demostraron que esta proteína disminuye los niveles de colesterol LDL, asociado a riesgo cardíaco.
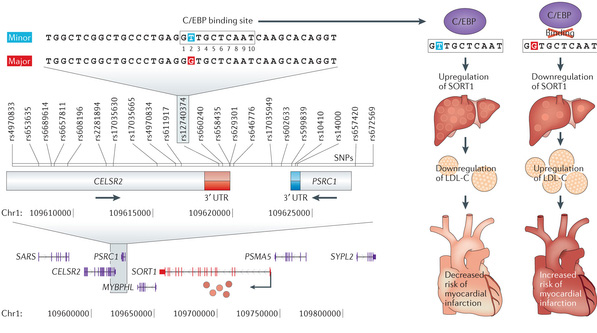
Otros ejemplos de eQTLs recientemente descubiertos han contribuido a explicar la asociaciones de variantes genéticas con enfermedades como el asma infantil, la enfermedad de Crohn o el lupus.
Este post ha sido realizado por Guillermo Peris (@Waltzing_piglet) y es una colaboración de Naukas con la Cátedra de Cultura Científica de la UPV/EHU.
Referencias
- Gene expression. Scitable (Nature Education).
- The role of regulatory variation in complex traits and disease. Albert, F.A. and Kruglyak, L. (2015) Nature Reviews Genetics 16, 197–212. doi:10.1038/nrg3891
- Expression quantitative trait loci: present and future. Nica A.C. and Dermitzakis E.T. (2013). Philos Trans R Soc Lond B Biol Sci.;368: 20120362
doi: 10.1098/rstb.2012.0362 - The study of eQTL variations by RNA-seq: from SNPs to phenotypes. Majewski, J. and Pastinen, T. (2011) Trends in Genetics 27 (2) , 72-79. doi: 10.1016/j.tig.2010.10.006.