Límite de resolución taxonómico
El uso de lentes como los prismáticos para poder observar la lejanía con más detalle fue un gran avance. Poder diferenciar algo peligroso de algo útil desde varios metros de distancia, de una forma rápida y efectiva, fue sin duda de una enorme utilidad. De la misma forma este hecho podría ayudarnos a comprender la revolución que supuso en el mundo de la identificación filogenética bacteriana el gen ADNr 16S.
Las bacterias, como el resto de los seres vivos, se organizan en distintos taxones. Al principio la clasificación y el ordenamiento se realizaba por las características fisiológicas y morfológicas. Podéis imaginar lo complejo y a veces, poco preciso, de la técnica. Clasificar atendiendo a la morfología de las bacterias, o al aspecto de sus colonias es muy complejo. No son animales, o plantas llenas de detalles visuales que analizar. Suelen ser de morfologías muy simples, y puede que los pigmentos o estructuras de sus colonias obedezcan más a una adaptación al entorno que a caracteres válidos para clasificar. Por otro lado, observando qué nutrientes consume un microorganismo o qué sustancias produce podemos tener datos algo mejores pero, qué hacer cuando sabemos que una misma especie, puede ser un patógeno mortal unas veces y otras un aliado indispensable de nuestra flora?
Las bacterias son propensas a lo que se conoce como: “transferencia horizontal”, consiste básicamente en captar material genético ajeno. Si una bacteria se hace con un trozo de ADN que contiene el gen para producir cierta sustancia, y hemos usado esa sustancia en nuestra clasificación, las cosas se pueden complicar. Por suerte, los taxónomos hacían uso de un gran número de características, por lo que aún sin técnicas modernas lograron hacerlo bastante bien.

Las cosas cambiaron cuando Carl Woese y sus colegas, en 1969 decidieron examinar el ARNr 16S, parte de la estructura del ribosoma bacteriano, la cual tiene varias características interesantes y muy útiles a la hora de usarse como “reloj molecular”. Tiene regiones en el gen que lo codifica que están bastante conservadas, no es un gen con tendencia a pasar por transferencia horizontal entre distintas bacterias y además tiene un tamaño bastante adecuado para su uso. Todo esto lo convirtió en una herramienta apropiada para la taxonomía microbiana.
El uso del gen que codifica el ARNr (ADNr) permitió separar a los procariotas en dos Dominios el Dominio Archaea y el Dominio Bacteria. Y concretamente el uso del gen ADNr 16S fue llave de una gran revolución taxonómica. Se pasó de complicadas pruebas fisiológicas y morfológicas a hacer una simple PCR, toda bacteria que mostrase una similitud menor de 97% era considerada una especie nueva.
La potencia de la prueba no se quedaba en el hecho de acelerar y facilitar el trabajo evitando, a veces, tediosas pruebas que tardaban meses. Además permitía estudiar bacterias incapaces de crecer en condiciones de laboratorio, abriendo enormemente el abanico de posibilidades.
Hablábamos al inicio del uso de prismáticos para observar objetos lejanos. Pongamos un ejemplo a modo de analogía siguiendo esa línea. El análisis del gen ADNr 16S ha sido el equivalente a usar unos prismáticos en observación de aves. Imaginad que en lugar de andar buscando plumas, egagrópilas, y otros restos de las aves, pudieseis verlas desde lejos, sin asustarlas. En lugar de meses para identificar una población podría hacerse en días. Una técnica increíble que… tiene problemas.
Continuemos con el ejemplo de los prismáticos antes de entrar más en el tema. Tenemos a nuestro observador de aves en el bosque, desde la lejanía es posible que surjan problemas, incluso equipado con unos buenos prismáticos. Es posible que no sea experto en un grupo concreto de aves, lo que puede llevarle a pensar que miembros de la misma especie son especies distintas. O puede que incluso siendo un gran experto, al mirar vea iguales muchas aves que en realidad son distintas. Estos son básicamente los problema que nos encontramos al usar el análisis del ANDr 16S.
Podemos encontrarnos ante un género lleno de especies distintas que quizás son cepas de una misma especie, o quizás ante la situación contraria. Tener varias cepas de una especie que en realidad son especies distintas. El primer caso a veces se debe a la prisa o el desconocimiento en la técnica de análisis moleculares. Cuando secuenciamos un gen para usarlo de guía filogenética es importante tratarlo adecuadamente, el secuenciado nos ofrecerá una ristra de nucleótidos, lo que viene a ser un código compuesto por 4 letras (ATCG), y otras tantas letras más que corresponden a dudas o problemas en la pauta de lectura. A estas dudas se las conoce como “bases degeneradas”.

Estas bases son básicamente la consecuencia de no poder resolver bien qué hay en cierto hueco del gen. Volviendo al ejemplo de nuestro ornitólogo de campo, sería como dudar del color del plumaje. ¿Era azul?, ¿era verde? ¿O era algo intermedio?¿Es adenina?, ¿guanina? ¿O quizás citosina?
Un caso aún peor es el de las N, las N aparecen en las lecturas como bases indeterminadas…pueden ser cualquier cosa o no ser nada. Las secuencias subidas a las bases de datos como secuencias definitivas que contienen muchas N (indeterminaciones) son una enorme fuente de problemas para futuros estudios.
Así que estamos ante un serio problema, puede que no hiciéramos bien el experimento y por eso nuestra lectura está llena de dudas, puede que nuestra bacteria tenga “complicaciones” en su estructura genética que impiden una lectura satisfactoria. En cualquier caso, son demasiadas las ocasiones en las que esto no se tiene en cuenta, y directamente las secuencias se cargan en las bases de datos, asumiendo que son correctas. En otros casos, el trabajo se hace bien, pero la técnica es insuficiente. Para el ejemplo del ornitólogo estaríamos ante un experto que trata de diferenciar aves muy parecidas desde lejos. Por muy bueno que sea, a cierta distancia las aves son exactamente iguales. Lo mismo ocurriría con las bacterias, lo más probable es que muchas cepas diesen valores de más de un 99% de similitud siendo especies distintas.
El uso de las técnicas moleculares aplicadas al genoma ha ayudado mucho, pero también ha ido en detrimento de otras técnicas clásicas, que sin desplazarlas del todo si que han quedado en un segundo plano. No podemos volver atrás y darle más importancia a características moleculares que a secuencias de genes importantes. Pero si podemos utilizarlas de una forma más cooperativa. El ornitólogo puede buscar restos en los nidos tras hacer sus observaciones desde lejos. Igual se pueden comprobar parámetros como perfiles de fragmentos repetitivos en el genoma, comportamiento fisiológico, etc… Así si una secuencia nos indica que estamos ante lo mismo, pero todo lo demás nos indica lo contrario, podremos plantearnos que el ADNr 16S no tiene la suficiente resolución para discriminar.
Todo esto puede parecer un problema trivial, quizás no debería importarnos mucho que se llame a una bacteria con un nombre u otro. Sin embargo algunos estudios (Moore, et al 2010) cuentan como por ejemplo algunas enfermedad infecciosas causadas por un microorganismo que no sea correctamente identificado a partir de un aislamiento a nivel de especie conduce directamente a relacionar este microorganismo con patogenicidad , riesgo de contagios. Implicación en diagnósticos , tratamientos y estrategias para la prevención de la propagación y erradicación de dicho patógeno… Vamos, que los riesgos inherentes a los microorganismos están directamente vinculados a los nombres de especies. Y, por supuesto , los niveles de riesgos biológicos asociados a nombres están directamente relacionadas con todos los aspectos de seguridad de la biotecnología y la bioseguridad .
Podemos ver un ejemplo claro de esto con Burkholderia (Peter Vandamme et al, 2014 ).
Burkholderia es un género con especies que tienen bastante actividad beneficiosa en plantas, sin embargo también despierta muchas preocupaciones por tener especies pertenecientes a grupos patógenos (Parke et al, 2001 ). Varias especies que están dentro de grupos beneficiosos para plantas han estado involucradas en infecciones humanas. De entre estas especies, especialmente Burkholderia fungorum ha sido aislada de una amplia gama de muestras humanas y veterinarias incluyendo: sangre, fluido cerebro-espinal humano , secreciones vaginales , encéfalo de cerdos con déficit neurológico, esputo y muestras de lavado de pacientes con fibrosis quística. También de lesiones del tallo cerebral de un ciervo , y de la nariz de varios ratones. Todo esto no supone un buen curriculum para una bacteria que pretende ser beneficiosa para la agricultura.
Como veis, la mala identificación si que puede llegar a ser un problema. Durante estos últimos años he estado trabajando con una bacteria perteneciente al género Methylobacterium. Este género es especialmente dado a problemas, contiene muchos reordenamientos de varias especies inicialmente mal clasificadas, y por si fuera poco las bases de datos están cargadas de secuencias mal corregidas o como mínimo sospechosas. Cuando tuvimos las secuencias correspondientes al gen ADNr 16S de dos cepas que estudiábamos decidí comprobar cómo encajarían dentro de un árbol filogenético construido con todas las secuencias completas del gen contenidas en las bases de datos oficiales. Y me llevé una gran sorpresa…

Imaginad que ocurre algo similar a Burkholderia, y que aparece un patógeno importante asociado a Methylobacterium radiodurans. Cada vez que alguien en un hospital realice una PCR buscando comprobar quién es el causante de la enfermedad dentro del género Methylobacterium…imaginad cuantas posibilidades tiene de caer cerca de M. radiodurans. Y obviamente, no es normal que una bacteria esté repartida por tantas zonas de un árbol filogenético.
Para seguir con la investigación, decidimos limpiar el árbol de todas las secuencias extrañas y llenas de indeterminaciones. Obtuvimos un árbol bastante más claro, pero que seguía teniendo alguna cosa extraña. Estábamos ante el límite de resolución de la prueba.
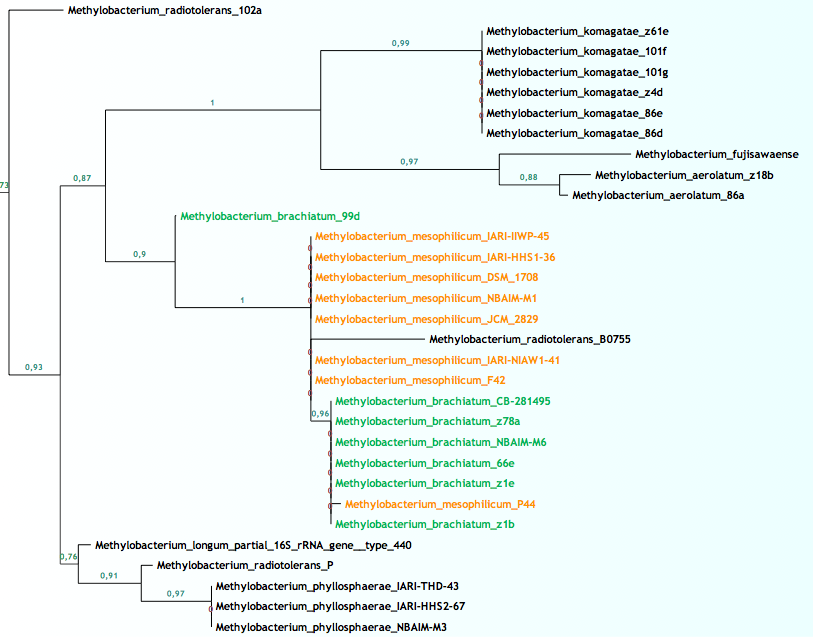
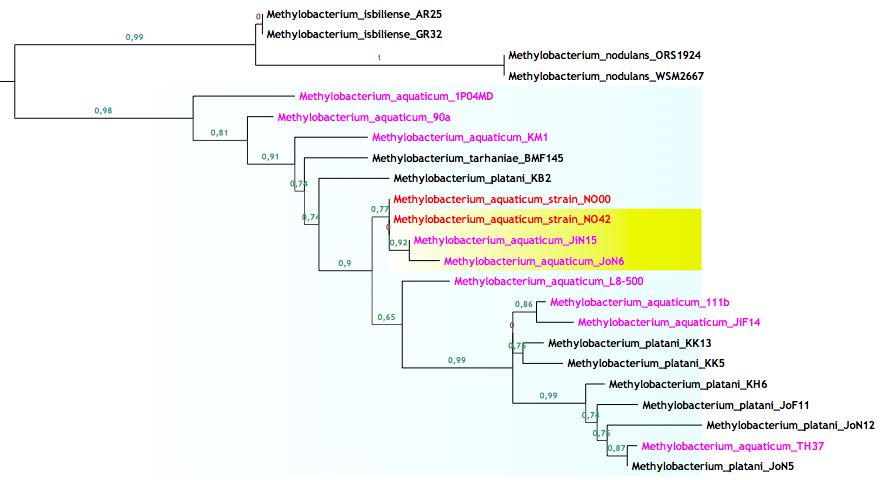
En las figuras de arriba podemos ver ramas del árbol filogenético del género Methylobacterium para el gen ADNr 16S, resuelto con las últimas cepas y procesado con las últimas técnicas. La longitud de las ramas indica la cercanía entre cada cepa y los números hablan de las posibilidades de que esa sea la correcta ordenación. La figura 4 muestra como varias especies distintas tienen una similitud máxima entre sus secuencias, lo que las coloca en la misma rama, mientras que en la figura 5 vemos lo contrario, una especie que posee cepas con grandes diferencias entre sus secuencias.
Básicamente la explicación para los distintos nombres en la figura 4 se debe a que además del gen ADNr 16S se han utilizado otros genes añadiendo resolución a la clasificación. Sin embargo en la rama de la figura 5 tan sólo hemos usado un gen, el gen por excelencia que poco a poco va perdiendo su omnipotencia.
Como veis, es importante mejorar las técnicas de clasificación filogenéticas. Y curiosamente para darnos cuenta de sus limitaciones a veces tenemos que usar técnicas más clásicas. El uso de varios genes altamente conservados en una especie se conoce como MLSA (Multi Locus Sequence Analysis) y viene a ser el telescopio del ornitólogo o esos grupos de radiotelescopios que permiten discriminar qué son estrellas binarias y qué son súper gigantes rojas. Es complicado encontrar cuáles son los genes apropiados para estos MLSA en cada especie, pero es el siguiente paso en el mundo de la taxonomía microbiana. Algo más rápido que la hibridación completa y más seguro que las aproximaciones de la taxonomía numérica mediante caracteres fisiológicos y morfológicos.
Este post ha sido realizado por José Jesús Gallego Parrilla (@Raven_neo) y es una colaboración de Naukas con la Cátedra de Cultura Científica de la UPV/EHU.
Referencias y más información:
Moore, E.R.B., Mihaylova, S.A., Vandamme, P., Krichevsky,
M.I., and Dijkshoorn, L. (2010) Microbial systematics and taxonomy: relevance for a microbial commons. Res Micro-biol 161: 430–438.
Prokaryotic taxonomy in the sequencing era – the polyphasic approach revisitedemi_2615 291..317
Peter Kämpfer1* and Stefanie P. Glaeser2
1Institut für Angewandte Mikrobiologie und 2Institut für Mikro- und Molekularbiologie, Justus-Liebig-Universität Giessen, Heinrich-Buff-Ring 26, D-35392 Giessen, Germany.
Time to revisit polyphasic taxonomy Peter Vandamme • Charlotte Peeters
DOI: 10.13140/RG.2.1.2250.3528
Límite de resolución taxonó…
[…] El uso de lentes como los prismáticos para poder observar la lejanía con más detalle fue un gran avance. Poder diferenciar algo peligroso de algo útil desde varios metros de distancia, de una forma rápida y efectiva, fue sin duda de una enorme […]
La pregunta Naukas 2016 – JJ Gallego – Naukas
[…] Puede parecer una tontería, sobretodo porque la especie biológica es uno de los conceptos más básicos en biología. Sin embargo a poco que nos ponemos a mirarlo de cerca, empezamos a ver que tiene problemas y límites. Sobre todo cuando lo necesitamos en el mundo de la taxonomía microbiológica. Allí la definición más extendida de especie biológica deja de funcionar quedando sujeta a parámetros laxos que cambian casi tras cada gran congreso de taxónomos. Hablé un poco de ello en el Cuaderno de Cultura Científica. […]
¿Cómo una bacteria inofensiva de Gambia acaba generando una epidemia en Wisconsin? – Cuaderno de Cultura Científica
[…] Cerrada la vía del estudio en el medio ambiente, el esfuerzo se centró en la secuencia genética de las muestras obtenidas de los enfermos2. -Y Ahora empieza quizás la parte más complicada, pero si ha llegado hasta aquí leyendo, no se asuste estimado lector, porque vamos a intentar entender lo básico de algunas técnicas. (Si además queréis saber más de taxonomía bacteriana y sus limitaciones con técnicas de antes de ayer podéis leer mi colaboración en la Cátedra de Cultura Científica). […]