¿Cómo una bacteria inofensiva de Gambia acaba generando una epidemia en Wisconsin?
Elizabethkingia anophelis fue aislada en 2011 a partir de muestras del intestino medio del complejo de criptoespecies al que pertenece el mosquito Anopheles gambiae1. Aunque podría haber quedado olvidada entre las publicaciones de las revistas de taxonomía, la bacteria saltó a la fama en 2016 cuando las noticias de Wisconsin se hicieron eco de su existencia, algo que cuando hablamos de bacterias no suele estar relacionado con buenas nuevas. E. anophelis estaba tras la infección de más de 60 personas y lo que era peor, era la causa de la muerte de 20.
¿Qué hacia una bacteria sin historial como patógeno infectando y matando gente?, una bacteria que además, habitualmente vive en el sistema digestivo de mosquitos. Sin olvidar el dato climático, los mosquitos no son algo común en Wisconsin durante el invierno.
No es una sorpresa ver a microorganismos inofensivos convertidos en patógenos: los cambios en el genoma, los plásmidos y otros mecanismos son capaces de generar esta situación. Por ello el CDC (Centro de control de enfermedades) hizo algo poco común, liberó por twitter el genoma recién secuenciado e imagino que sin procesar, rápidamente algunos microbiólogos como Kat Holt o Sylvain Brisse se unieron para solucionar el problema.

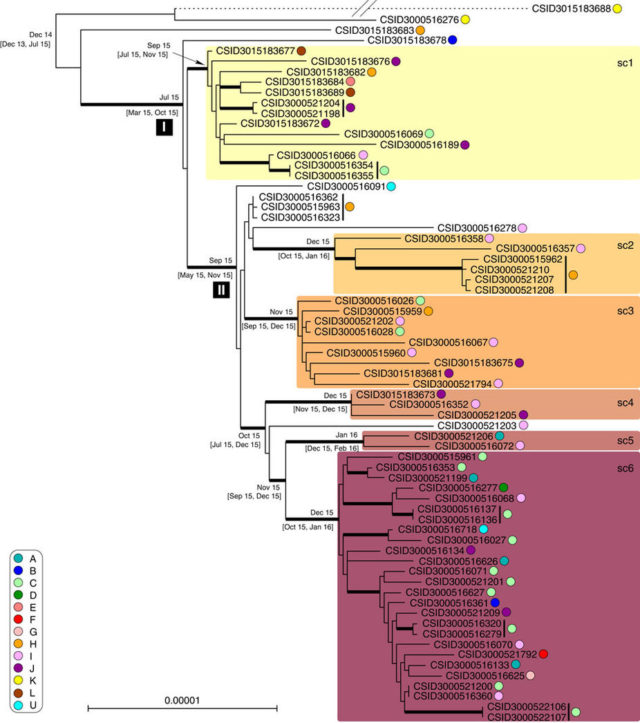
La primera cosa que observaron es que las secuencias procesadas de las muestras de los enfermos eran demasiado distintas. Normalmente las epidemias las causan una misma cepa con una secuencia genética casi idéntica con variaciones mínimas propias de cambios individuales, 40, 50 o incluso 100 pares de bases dentro del total. Sin embargo en esta ocasión las diferencias eran mucho mayores. Así que tras asegurarse por medio de herramientas mucho más potentes como el análisis cgMLST y aproximaciones comparativas de SNP en el genoma completo… Se llegó a la indudable conclusión de que aún siendo tan distintas, eran no sólo la misma especie sino que provenían de una misma cepa-ancestro. La diferencia entre ellas provenía principalmente de una enorme trozo insertado en el medio del gen mutY.
El gen mutY, se encarga de codificar para una proteína encargada de reparar errores en el ADN, así que ahora podéis imaginar que tras llevar un trozo de código más grande que la propia secuencia, la capacidad para producir la proteína desaparece completamente. Sin reparación, tenemos mutación descontrolada, y aunque la mayoría de las mutaciones son negativas o sin función…con el suficiente tiempo un pequeño porcentaje podrán dar habilidades no deseadas (por nosotros) a las bacterias. Y claro, de camino también ayudaría a volver locos a los microbiólogos que trataban de identificar al patógeno.
Aunque resuelto e identificado el culpable, seguía sin estar claro cómo una bacteria de esas características había llegado allí y lo que era más extraño, el modo en el que aparecieron los casos. Normalmente las epidemias aparecen en un hospital o lugar concreto desde el que se expanden, pero en este caso apareció de forma diseminada. El CDC analizó todas las posibles fuentes de esta bacteria dentro del ambiente hospitalario sin dar en ningún comento con la bacteria en el medio. Por lo que el foco se centró en la comunidad.
Cerrada la vía del estudio en el medio ambiente, el esfuerzo se centró en la secuencia genética de las muestras obtenidas de los enfermos2. -Y Ahora empieza quizás la parte más complicada, pero si ha llegado hasta aquí leyendo, no se asuste estimado lector, porque vamos a intentar entender lo básico de algunas técnicas. (Si además queréis saber más de taxonomía bacteriana y sus limitaciones con técnicas de antes de ayer podéis leer mi colaboración en la Cátedra de Cultura Científica).
El árbol filogenético se realizó usando la que es quizás la herramienta más potente en taxonomía microbiona, el cgMLST. Esta técnica está basada en el análisis MLST, pero vayamos poco a poco. En taxonomía microbiana existen muchas aproximaciones a la descripción de especies. Centrándonos en los métodos moleculares, concretamente en los que implican secuencias genéticas, el más conocido es el análsis del ADNr 16S del que hablo en el texto citado en el párrafo anterior. Voy a contarlo de una forma muy sencilla, casi näif:
Si queremos describir en base a la secuencia genética necesitamos un código que sea muy similar, que cambie muy poco, y ya sabemos que los seres vivos tienden a mutar y cambiar con las generaciones. Pero existen genes muy muy importantes, tanto que los cambios en ellos son muy pocos. Estos genes, o mejor dicho trozos de ellos, son los candidatos elegidos para secuenciar e identificar mediante multilocus sequence typing (MLST). Cada especie bacteriana tiene un número de genes de este tipo, suelen elegirse siete y en base a ellos se realizan árboles mediante procesos estadísticos (en los que no vamos a entrar). Quizás os preguntéis por qué no usar Whole-genome sequencing (WGS), la secuenciación del genoma completo y la hibridación es el método “final” para publicar una nueva especie microbiana, básicamente alineas el genoma completo de esa especie y la más cercana para demostrar que hay partes que no son iguales y que esas partes son lo suficientemente grandes para considerarse una especie nueva. Sin embargo este método no es útil en epidemiología. Es complicado estandarizar, ya que arroja tantos por ciento de similitud, lo que complica un poco la colaboración entre distintos centros de investigación.
¿La solución?, ya la imagináis core genome MLST (cgMLST). Retrocedamos a los médicos que tomaban muestras de los enfermos, muestras de una bacteria que cambiaba casi cada día…bacterias que siendo responsables de la misma epidemia eran sensiblemente distintas entre ellas. El genoma de las bacterias estudiadas era distinto en gran parte, pero claramente perteneciente a la misma especie. A la suma de todos esos genomas que tenemos es a lo que llamamos pan-genoma,
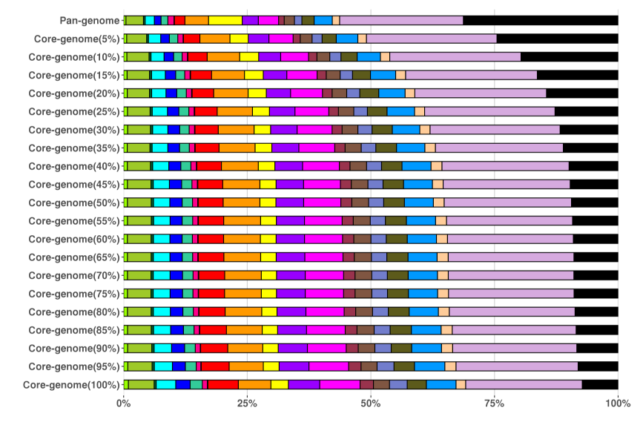
El Core Genoma sería (simplificando muchísimo) las partes de la secuencia que van caracterizando las similitudes entre cepas de la misma especie. Dejando los “genes únicos” como las zonas que diferencian a cada cepa de las demás. Así que tenemos un Pan-genoma que contiene partes compartidas llamadas Core-genoma, partes distintas llamadas genes únicos. El Pan-genoma por decirlo de otra forma, sería una secuencia consenso entre las cepas.
Pues mezclando ambos conceptos se desarrolló el cgMLST. En el cual se estandarizó y dotó de un sistema numérico a los alelos ayudándose del mapeo de single nucleotide polymorphism (SNP), que son pequeños cambios en el ADN usados también para diferenciar y clasificar. La ventaja de todo este sistema, es su velocidad y usabilidad una vez automatizado. Tomar una muestra de la bacteria, secuenciar los genes MLST, y cotejar con el sistema cgMLST para poder colocar y clasificar.
Se tomaron muestras casi diarias de prácticamente todos los pacientes, y tras procesarlas en el cgMLST se vio que el core-genoma era idéntico excepto por un único SNP encontrado en uno de los aislamientos (en el mismo paciente otros dos aislamientos también fueron distintos para ese SNP) Este dato confirmó lo dicho al inicio de la historia, que se trataba de una bacteria con un origen común pero con una gran capacidad de mutar.
Por si fuera poco sorprendente, los análisis bayesianos demostraron que había una “firma temporal”, lo que implicaba que la cepa de la epidemia se había continuado diversificando de una forma mesurable durante el curso de la epidemia. Se estimó que el ratio de evolución era de 5.98 × 10−6 substituciones nucleotídicas por año y sitio. Atendiendo al ratio de diversificación y a que las muestras fueron tomadas alrededor de julio de 2015. Se pudo determinar la aparición del ancestro de la cepa epidémica aproximaciones a finales de diciembre de 2014. Para asegurarse, los investigadores realizaron un experimento similar pero atendiendo a los SNP del genoma completo y no sólo de los cgMLST, este experimento situó el origen de la cepa en agosto de 2014. Uniendo ambos resultados se confirmó que la cepa apareció un año antes de la epidemia, y evolucionó en un reservorio desconocido para luego generar la epidemia, siguiendo su diversificación durante todo el proceso.
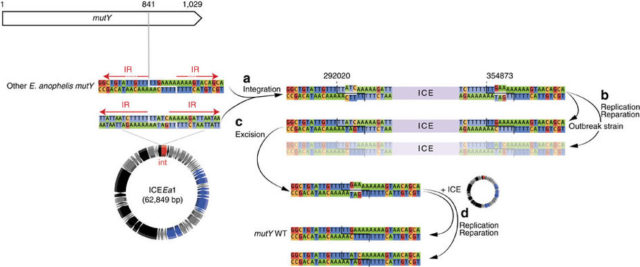
Estudiando el elemento externo que se había insertado en el genoma de la bacteria causando este fenómeno de “hipermutación” y patogenia, los investigadores descubrieron que se trataba del elemento, al que bautizaron como ICEEa1 (integrative and conjugative element 1 of E. anophelis). Era en realidad un enorme paquete que originalmente pertenecía a un Sistema de secreción tipo 4 de Bacteroidetes (T4SS-B). El ICEEa1, además de los 12 genes que codificaban para el T4SS, también incluía: un set completo de genes para integración/escisión y conjugación bacteriana, una relaxasa, una ATPasa, la metilasa de ADN N-6 y reguladores para tetraciclinas. Si esto fuese poco, también se encontraron genes para sistemas de producción de sideróforos (para capturar hierro libre) y bombas de transporte para: cobalto, zinc, cadmio, níquel, cobre y mercurio.
La incapacidad de reparar su genoma por tener todo esto en medio del mutY, parece que generó en la cepa original muchos daños por estrés oxidativo. El tipo de mutaciones así lo indicaban, así que observar esta increíble resistencia a metales pesados cuadraba bastante con la hipótesis de que la cepa inicial evolucionó en un ambiente contaminado en la comunidad y no en hospitales.
Aunque lo más sorprendente y preocupante fue descubrir que el ICEEa1 era de “quita y pon”, la bacteria podía llevar insertado el trozo y perder su capacidad para reparar el genoma, pero a cambio convertirse en fenotipo hipermutador, adquirir resistencia a metales pesados y volverse muy patógena. Y en cualquier momento soltar el trozo para recuperar sus capacidades normales manteniendo algunas de las adquiridas. No os imaginéis algo voluntario, más bien pensad en una enorme población de bacterias que van multiplicándose y por transferencia horizontal pasándose el trozo unas a otras generando estas cepas hipermutantes de vez en cuando.
Dicho esto, a los investigadores sólo les faltaba una cosa por contar, ¿por qué se había vuelto patógena ahora si nunca había causado problemas? Pues quizás siempre lo había sido… Como ya se ha dicho muchas veces la taxonomía basada en el ADNr16S y la tipificación por métodos bioquímicos no son por si mismas suficientes, esto es algo que empieza a admitirse lentamente. Los investigadores piensan que muchas de las bacterias identificadas como Elizabethkingia meningoseptica, un típico patógeno de ambientes hospitalarios eran en realidad E. anophelis, por lo que se propuso la actualización de las bases de datos del MALDI-TOF, herramienta usada en la identificación rápida de patógenos. Y se puso en aviso a la comunidad de epidemiólogos pues trozos como el ICEEa1 podían pasar a otras bacterias distintas dejando el aviso de que una nueva epidemia es sólo cuestión de tiempo.
Este post ha sido realizado por José Jesús Gallego Parrilla (@Raven_neo) y es una colaboración de Naukas con la Cátedra de Cultura Científica de la UPV/EHU.
Referencias científicas y más información:
1.Kämpfer, P. et al. Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. International Journal of Systematic and Evolutionary Microbiology 61, 2670–2675 (2011).
2.Perrin, A. et al. Evolutionary dynamics and genomic features of the Elizabethkingia anophelis 2015 to 2016 Wisconsin outbreak strain. Nature communications 8, 15483 (2017).