Proteómica: cuando las proteínas y la espectrometría de masas van de la mano
La Facultad de Ciencias de Bilbao comenzó su andadura en el curso 1968/69. 50 años después la Facultad de Ciencia y Tecnología de la UPV/EHU celebrará dicho acontecimiento dando a conocer el impacto que la Facultad ha tenido en nuestra sociedad. Durante las próximas semanas en el Cuaderno de Cultura Científica y en Zientzia Kaiera se publicarán regularmente artículos que narren algunas de las contribuciones más significativas realizadas a lo largo de estas cinco décadas. Comenzamos con la serie “Espectrometría de masas”, técnica analítica que supone el ejemplo perfecto del incesante avance de la Ciencia y la Tecnología.

La espectrometría de masas (MS) es una técnica habitual entre los químicos desde hace ya 100 años, pero no fue hasta hace 3 décadas que se empezó a utilizar como técnica de análisis de proteínas. Para analizar un analito mediante espectrometría de masas (MS) es necesario ionizar dicho analito y pasarlo a fase gaseosa. Durante mucho tiempo, la posibilidad de ionizar macromoléculas como las proteínas o fragmentos de proteínas, es decir péptidos, y pasarlos a fase gaseosa se consideraba una tarea tan posible como hacer volar a un elefante. Sin embargo, el desarrollo y comercialización de dos métodos de ionización a los que se denominó ionización “suave”, demostraron que era posible analizar macromoléculas del tamaño de las proteínas, tanto en disolución como en estado seco cristalino; éstos métodos de ionización se conocen hoy día como ionización electrospray (ESI) y ionización por desorción láser asistida por matriz, (matrix assisted laser desorption ionization) (MALDI). En ESI, se aplica un alto voltaje a la solución que contiene el analito a su paso por una fina aguja perforada. A medida que se va evaporando la solución con las moléculas cargadas, se va reduciendo el tamaño de las gotas, hasta conseguir que pasen a estado gaseoso. Por su parte, en MALDI se dispara con láser a una placa de acero en la que se ha secado el analito junto con una matriz. La matriz absorbe la energía del láser que a su vez se transfiere a la muestra, haciendo que cambie a estado gaseoso.
Pero, ¿por qué las proteínas? ¿a qué viene ese interés por analizarlas? La razón es que las proteínas son las moléculas fundamentales de todos los seres vivos. Las proteínas son las encargadas de realizar diversas tareas en nuestras células; entre otras, catalizan reacciones químicas, transportan moléculas, mantienen la estructura celular y nos protegen frente a los patógenos. Nuestro cuerpo está formado por trillones de células, y cada una de ellas cuenta con miles de proteínas diferentes. De hecho, dependiendo del catálogo o conjunto de proteínas que contenga un tipo de célula, llevará a cabo unas u otras funciones y tendrá unas u otras características. Todas las células de un ser vivo contienen la misma información genética, es decir, el genoma de todas sus células es idéntico. La información necesaria para producir cada proteína está escrita en los genes. Dicho de otro modo, los genes son las recetas que se siguen para crear las proteínas, y dependiendo de las recetas que se utilicen en cada momento, se producirán unas proteínas u otras. Se denomina proteoma a la colección de proteínas de las que dispone una célula en un momento concreto. A diferencia del genoma, el proteoma celular varía continuamente; en respuesta a cualquier estímulo que llegue a las células, o cuando deben hacer frente a cualquier ataque, es decir, con el fin de adaptarse al entorno, las células producirán diferentes proteínas que trabajarán conjuntamente. Esto explica cómo, contando ambas con el mismo genoma, una oruga se convierta en mariposa (Imagen 1).

Las proteínas, al igual que nosotros, tienen sus propias «redes sociales». Cumplen sus funciones interaccionando entre ellas más que individualmente. A pesar de ello, estas interacciones suelen ser, en la mayoría de los casos, temporales. Se crean y se destruyen según las necesidades celulares. Habiendo millones de proteínas en la células, la red de interacciones intracelulares es tan compleja como la que puede haber entre todas las personas del planeta. El funcionamiento de esa red de proteínas condicionará la salud de la célula y, en consecuencia, la de todo el organismo. Por eso es tan importante estudiar las proteínas y sus interacciones, es decir, los proteomas, e intentar interpretarlos en su conjunto. En consecuencia, uno de los retos fundamentales de la biología actual es describir los proteomas cualitativamente y cuantitativamente. Dicho objetivo parece alcanzable y pocos dudan del liderazgo que ha tomado la proteómica en este camino.
La proteómica estudia los proteomas. Fue en 1975 cuando por primera vez se separaron miles de proteínas de la bacteria Escherichia coli mediante electroforesis bidimensional en gel (2DE). Sin embargo, la identificación de dichas proteínas, concretar cuáles eran exactamente, constituía otro reto diferente. Para abordarlo, fue necesario, tanto desarrollar técnicas de secuenciación parcial, léase degradación de Edman, como ajustar la propia técnica de espectrometría de masas (MS) al análisis de proteínas. La MS es, a día de hoy, el método que se utiliza habitualmente para identificar proteínas. Para poder interpretar los proteomas en su conjunto, además de identificar las proteínas, hay que cuantificarlas. Los primeros pasos para la cuantificación de proteínas se dieron mediante el análisis de imágenes de los geles de 2DE. Si bien sigue siendo una técnica adecuada para el análisis proteómico, hoy en día ocupa un segundo lugar, muy por detrás de la proteómica basada en la espectrometría de masas (MS-based proteomics). De hecho, actualmente, la MS es el método habitual para identificar y cuantificar las proteínas en muestras proteicas complejas. Se ha convertido pues en una técnica fundamental para interpretar la información codificada en el genoma.
Tal y como se ha mencionado anteriormente, las técnicas habituales utilizadas para ionizar y evaporar proteínas y péptidos son ESI y MALDI. La técnica MALDI-MS se utiliza para analizar mezclas de péptidos relativamente simples. En cambio, los sistemas ESI-MS acoplados a la cromatografía líquida (LC-MS), son los más adecuados para el análisis de muestras complejas. Siendo los proteomas, por definición, complejos, las técnicas de LC-MS han prevalecido en la proteómica. La precisión, sensibilidad y resolución de masas, junto con su velocidad, son las claves del éxito de las técnicas de LC-MS. Durante los últimos años, se ha podido identificar, cuantificar y caracterizar un enorme número de proteomas mediante diferentes procedimientos de LC-MS. Entre los logros conseguidos a nivel de proteómica, cabe destacar dos borradores del proteoma humano publicados en 2014, y el actual Proyecto Proteoma Humano (HPP) dirigido por la Organización Mundial del Proteoma Humano (HUPO)
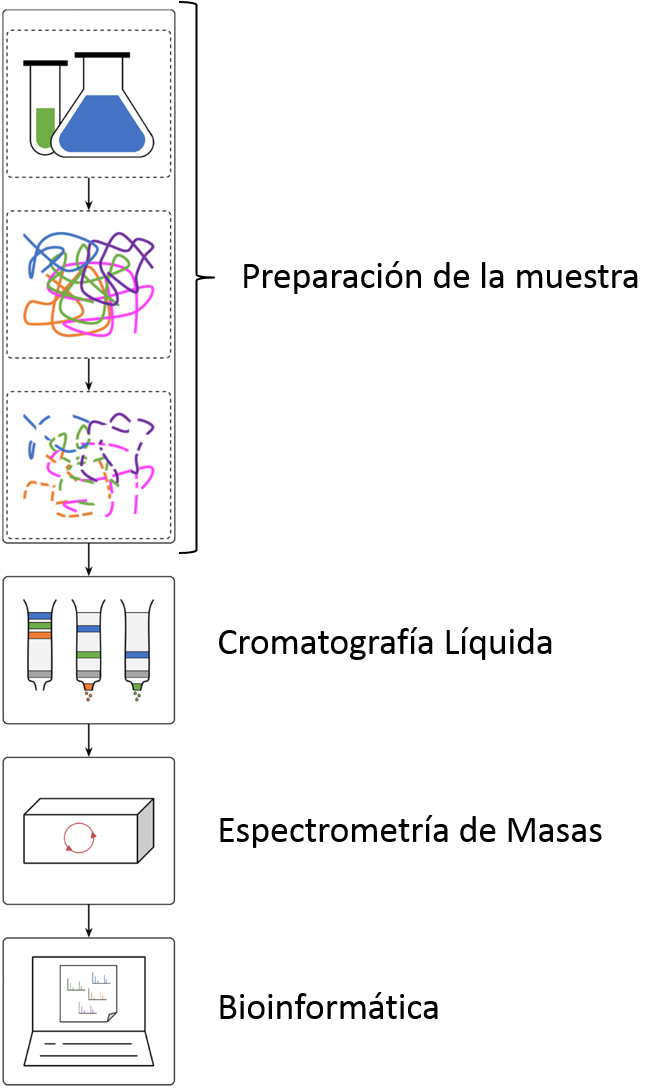
Pero, ¿qué es lo que hacemos exactamente en un laboratorio de proteómica? Sea cual sea la muestra recibida (muestra obtenida de una biopsia, células animales o vegetales, bacterias, secreción biológica…), habitualmente se aplica el flujo de trabajo denominado proteómica bottom-up o proteómica ascendente, de abajo hacia arriba (Imagen 2). En primer lugar, se extraen las proteínas de la muestra. A continuación, se utiliza una enzima que digiere las proteínas, habitualmente la tripsina, para trocear las proteínas en trozos pequeños denominados péptidos. Los péptidos de la mezcla se separan en la cromatografía líquida de fase inversa acoplada a la ionización por electrospray. A medida que los péptidos eluyen de la columna, se van ionizando y pasan al espectrómetro de masas. Allí, los iones de los péptido se van separando y detectando en función de su relación masa-carga (m/z). Los péptidos más abundantes se aíslan del resto, y se fragmentan, dando lugar a espectros de fragmentación. Estos espectros son los que contienen la información que permite identificar y cuantificar los péptidos. Finalmente, los datos obtenidos por el espectrómetro de masas se analizan mediante herramientas informáticas específicas. De esta manera, es posible saber qué proteínas hay en la muestra analizada y en qué cantidad.
Durante la última década, la proteómica basada en la MS se ha convertido en una de las principales herramientas analíticas para las biociencias. Ha permitido estudiar las proteínas desde diversas vertientes, y ha facilitado el camino para conocer sus estructuras, variaciones, cantidades, modificaciones postraduccionales e interacciones. Además, gracias a la proteómica nos encontramos más cerca de identificar las proteínas implicadas en el desarrollo de diversas enfermedades y los biomarcadores necesarios para el diagnóstico de las mismas. Durante los próximos años, se irán completando los catálogos proteicos de diferentes condiciones celulares. Junto a los resultados obtenidos a partir de la genómica, epigenómica, metabolómica y otras «ómicas» a gran escala, los descubrimientos obtenidos gracias a la proteómica contribuirán a la construcción de modelos celulares. De hecho, esa es la tendencia actual: trabajar desde un punto de vista multidisciplinar con el fin de construir modelos matemáticos y estadísticos que ayuden a aclarar la complejidad de los procesos biológicos; y es precisamente ahí donde la proteómica jugará un papel relevante.
Más información
- Fenn JB (2003) “Electrospray wings for molecular elephants (Nobel lecture)” Angew Chem Int Ed Engl. 42(33), 3871-94. DOI:10.1002/anie.200300605
- Fenn JB et al. (1989) “Electrospray ionization for mass spectrometry of large biomolecules” Science 246(4926), 64-71. DOI: 10.1126/science.2675315
- Tanaka K et al. (1988) “Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry” Rapid Communications in Mass Spectrometry 2(8), 151–153. DOI:10.1002/rcm.1290020802
- Karas M and Hillenkamp F (1988) “Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons” Anal Chem. 60(20), 2299-301. DOI: 10.1021/ac00171a028
- O’Farrell (1975) “High resolution two-dimensional electrophoresis of proteins” J Biol Chem 250(10), 4007-21.
- Kim MS et al. (2014) “A draft map of the human proteome” Nature 509(7502), 575-81. DOI: 10.1038/nature13302
- Wilhelm M et al. (2014) “Mass-spectrometry-based draft of the human proteome” Nature 509(7502), 582-7. DOI: 10.1038/nature13319
- HUPO: https://hupo.org/human-proteome-project
Sobre los autores: Miren Josu Omaetxebarria, Nerea Osinalde, Jesusmari Arizmendi y Jabi Beaskoetxea, miembros del departamento de Bioquímica y Biología Molecular, y Kerman Aloria, técnico de SGIker.

.jpg){kind=link}
{kind=link}
El estudio de las proteínas – hell1p
[…] marcadores fluorescentes, la ultracentrifugación analítica, el esparcimiento dinámico de luz, la espectrometría de masas (MS), entre otros. Los resultados de estos estudios permiten proponer modelos de los mecanismos con […]